![]()
![]()
![]()
Alignment based Quantification
Disclaimer this tool needs a peptide database, quantified peptides and a list of peptides deduced by alignment.
Given an MS run in the mzLite or mzml format and a list of a list of peptides deduced by alignment., this tool iterates accross all and performs an XIC extration and quantification in similar to the PSMbasedQuantification tool.
One of the drawbacks of data-dependent acquisition is the stochastic nature of peptide ion selection for MSMS fragmentation as a prerequisite for peptide identification and quantification. A way to overcome this drawback is the transfer of identified ions from one run to another using the assumption that the run is merely lacking a successful MSMS scan, but still containing the peptide itself.
For each peptide ion the tools uses the scan time prediction derived using the quant based alignment tool to extract a XIC. To refine the derived scan time estimate, we then locally align the extracted XIC to the XIC of the aligned peptide using dynamic time warping.
Using this scan time estimate, we use wavelet based peak detection techniques to identify all peaks present in the XIC and select the most probable peak as our target for quantification. Using parameter estimation techniques we subsequently use peak fitting to fit a set of two gaussian models to the detected peak, from whom the one with the better fit is selected. This allows us not only to report how well the signal fitted to the theoretical expected peak shape but also to obtain accurate estimates for the peak area, our estimator for peptide ion abundance.
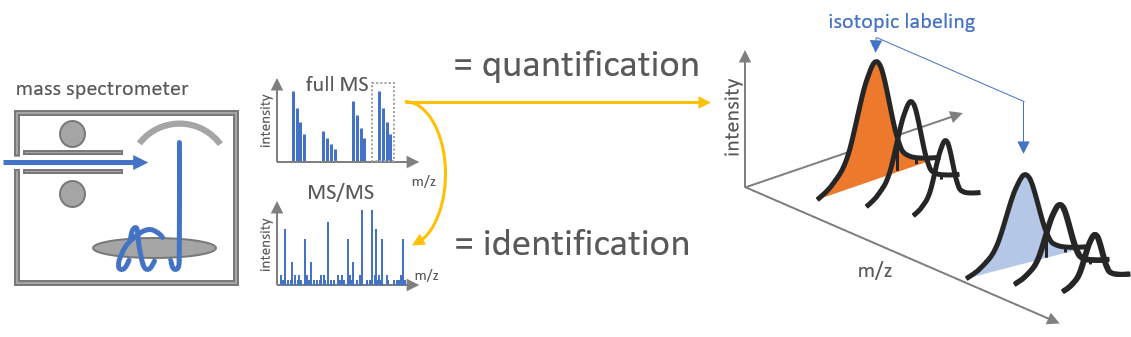
The quantification tool was designed to allow label-free quantification as well as quantification of full metabolic labeled samples. For this we use the known identity of one of the the peptide ions and calculate the m/z of the unobserved differentially labeled counterpart to extract and quantify the corresponding XIC.
Parameters
The following table gives an overview of the parameter set:
Parameter |
Default Value |
Description |
|---|---|---|
PerformLabeledQuantification |
true |
Indicates if a labeled quantification should be performed |
PerformLocalWarp |
true |
Indicates if dynamic time warping based scan time refinement should be performed |
XicExtraction |
{TopKPSMs = None; ScanTimeWindow = 2.; MzWindow_Da = 0.07; XicProcessing = Wavelet waveletParams} |
Parameters to tune the Xic extraction |
BaseLineCorrection |
Some { MaxIterations = 10; Lambda = 6; P = 0.05 } |
optional parameter, defines if and how baseline correction should be performed |
Parameter Generation
Parameters are handed to the cli tool as a .json file. you can download the default file here, or use an F# script, which can be downloaded or run in Binder at the top of the page, to write your own parameter file:
#r "nuget: BioFSharp.Mz, 0.1.5-beta"
#r "nuget: ProteomIQon, 0.0.1"
open ProteomIQon
open ProteomIQon.Domain
open FSharp.Stats.Signal
open BioFSharp.Mz
let defaultQuantificationParams :Dto.AlignmentBasedQuantificationParams =
///
let waveletParams :WaveletParameters =
{
Borderpadding = None
BorderPadMethod = Padding.BorderPaddingMethod.Random
InternalPaddingMethod = Padding.InternalPaddingMethod.LinearInterpolation
HugeGapPaddingMethod = Padding.HugeGapPaddingMethod.Zero
HugeGapPaddingDistance = 100.
MinPeakDistance = None
MinPeakLength = Some 0.1
MaxPeakLength = 1.5
NoiseQuantile = 0.01
MinSNR = 0.01
}
let XicExtraction =
{
TopKPSMs = None
ScanTimeWindow = 2.
MzWindow_Da = 0.07
XicProcessing = Wavelet waveletParams
}
let BaseLineCorrection =
{
MaxIterations = 10
Lambda = 6
P = 0.05
}
{
PerformLabeledQuantification = true
PerformLocalWarp = true
XicExtraction = XicExtraction
BaseLineCorrection = Some BaseLineCorrection
}
let serialized =
defaultQuantificationParams
|> Json.serialize
Executing the Tool
Disclaimer this tool needs a peptide database, quantified peptides and a list of peptides deduced by alignment.
proteomiqon-alignmentbasedquantification -i "path/to/your/run.mzlite" -ii "path/to/your/run.align" -iii "path/to/your/run.alignmetric" -iv "path/to/your/run.quant" -d "path/to/your/database.sqlite" -o "path/to/your/outDirectory" -p "path/to/your/params.json"
It is also possible to call the tool on a lists of MS and scored psm files. If you have a multicore cpu it is possible to score multiple runs in parallel using the -c flag:
proteomiqon-alignmentbasedquantification -i "path/to/your/run1.mzlite" "path/to/your/run2.mzlite" -ii "path/to/your/run1.align" "path/to/your/run2.align" -iii "path/to/your/run1.alignmetric" "path/to/your/run2.alignmetric" -iv "path/to/your/run1.quant" "path/to/your/run2.quant" -d "path/to/your/database.sqlite" -o "path/to/your/outDirectory" -p "path/to/your/params.json" -c 2
By default files get matched by their position in the list. To perform a name based file match set the -mf flag:
proteomiqon-alignmentbasedquantification -i "path/to/your/run1.mzlite" "path/to/your/run2.mzlite" -ii "path/to/your/run1.align" "path/to/your/run2.align" -iii "path/to/your/run1.alignmetric" "path/to/your/run2.alignmetric" -iv "path/to/your/run1.quant" "path/to/your/run2.quant" -d "path/to/your/database.sqlite" -o "path/to/your/outDirectory" -p "path/to/your/params.json" -c 2 -mf
A detailed description of the CLI arguments the tool expects can be obtained by calling the tool:
proteomiqon-alignmentbasedquantification --help
from ProteomIQon
namespace FSharp
--------------------
namespace Microsoft.FSharp
from ProteomIQon
module AlignmentBasedQuantificationParams
from ProteomIQon.Dto
--------------------
type AlignmentBasedQuantificationParams =
{ PerformLabeledQuantification: bool
PerformLocalWarp: bool
XicExtraction: XicExtraction
BaseLineCorrection: BaseLineCorrection option }
from FSharp.Stats.Signal
| Random
| Zero
| Random
| NaN
| Delete
| Zero
| LinearInterpolation
| Random
| NaN
| Delete
| Zero
| LinearInterpolation
val XicExtraction : XicExtraction
--------------------
type XicExtraction =
{ ScanTimeWindow: float
MzWindow_Da: float
XicProcessing: XicProcessing
TopKPSMs: int option }
| SecondDerivative of SecondDerivativeParams
| Wavelet of WaveletParameters
union case XicProcessing.Wavelet: WaveletParameters -> XicProcessing
--------------------
module Wavelet
from FSharp.Stats.Signal
val BaseLineCorrection : BaseLineCorrection
--------------------
type BaseLineCorrection =
{ MaxIterations: int
Lambda: int
P: float }
from ProteomIQon