![]()
![]()
![]()
Peptide Spectrum Matching
Disclaimer this tool needs a peptide database to query against, if you did not create one yet you can do so by using the PeptideDB tool.
An established method to identify acquired MS/MS spectra is the comparison of each spectrum with peptides in a reference database.
Given raw a MS run in the mzLite or mzml format, this tool iterates accross all recorded MS/MS scans and determines the charge state of precursor ions which were selected for fragmentation. With this it is possible to query the peptide data base for every precursor ion mass +/- a tolerance (which defines the so called 'search space') and retrieve peptides that are theoretical candidates for a match. For each of the peptide candidates we create an theoretical spectrum in silico and compare it to the measured MS/MS scan.
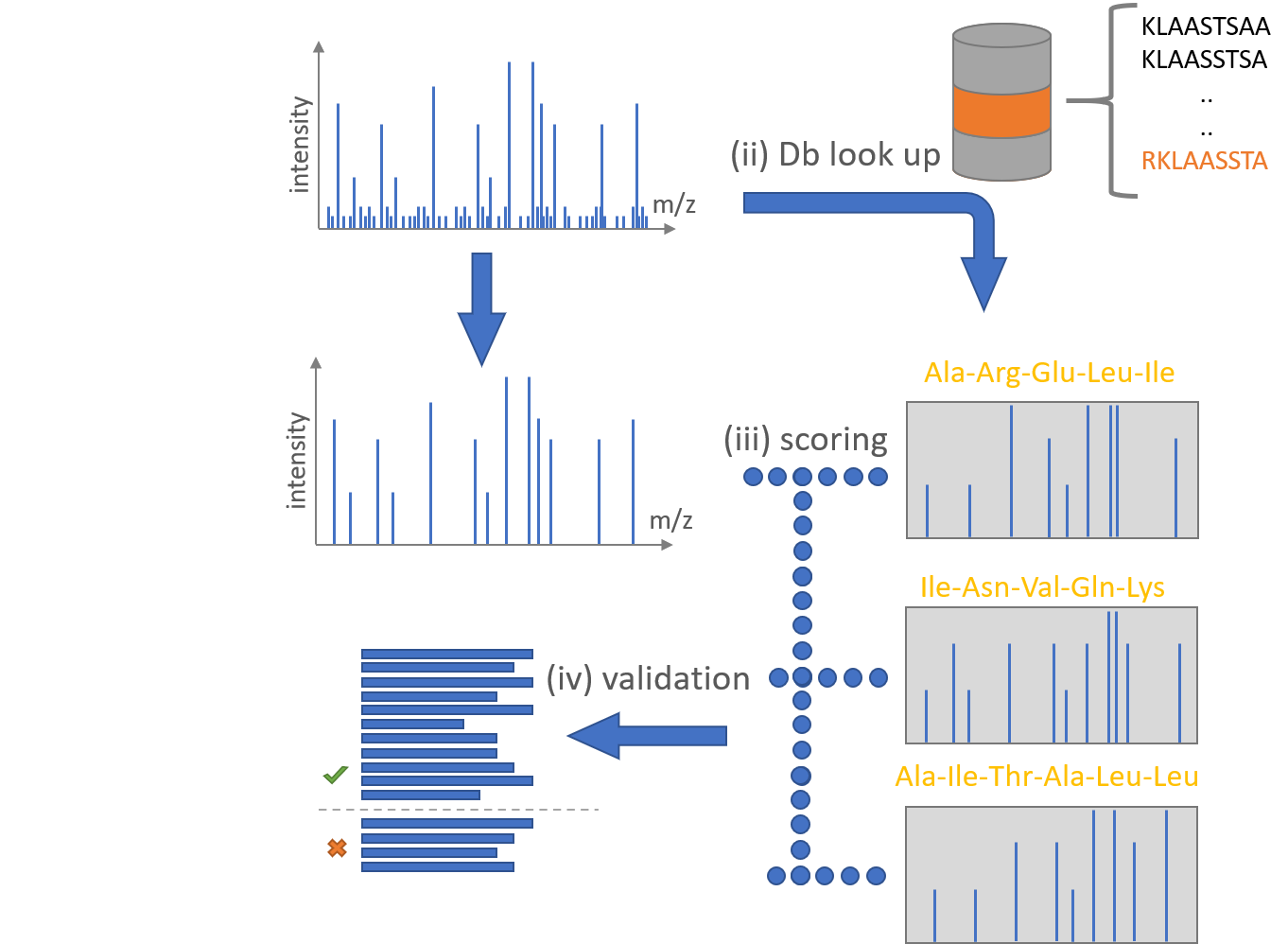
To measure similarity we use our own implementations of three established search enginge scores: SEQUEST, Andromeda and XTandem. The search space is extended by so called decoys. Decoys are reversed counterparts of peptides within the search space and allow us to assign a false discovery rate to each scored peptide using the PSMStatistics tool.
Parameters
The following table gives an overview of the parameter set:
Parameter |
Default Value |
Description |
|---|---|---|
ChargeStateDeterminationParams |
{ExpectedMinimalCharge = 2; ExpectedMaximumCharge = 5; Width = 1.1; MinIntensity = 0.15; DeltaMinIntensity = 0.3; NrOfRndSpectra = 10000} |
Parameters used for the charge state determination of the peptides |
LookUpPPM |
30 |
Mass range in Da in which potential peptides are selected |
MS2ScanRange |
100.,2000. |
m/z range for MS2 spectra |
nTerminalSeries |
NTerminalSeries.B |
Considered ions starting from the N-Terminus |
cTerminalSeries |
CTerminalSeries.Y |
Considered ions starting from the C-Terminus |
Andromeda |
{PMinPMax = 4,10; MatchingIonTolerancePPM = 100.} |
Andromeda scoring parameters |
Parameter Generation
Parameters are handed to the cli tool as a .json file. you can download the default file here, or use an F# script, which can be downloaded or run in Binder at the top of the page, to write your own parameter file:
#r "nuget: BioFSharp.Mz, 0.1.5-beta"
#r "nuget: Newtonsoft.Json, 12.0.3"
#r "nuget: ProteomIQon, 0.0.5"
open BioFSharp.Mz.SearchDB
open Newtonsoft.Json
open ProteomIQon
open ProteomIQon.Domain
open BioFSharp.Mz
let chargeDetermParams :ChargeState.ChargeDetermParams =
{
ExpectedMinimalCharge = 2
ExpectedMaximumCharge = 5
Width = 1.1
MinIntensity = 0.15
DeltaMinIntensity = 0.3
NrOfRndSpectra = 10000
}
let andromedaParams: AndromedaParams =
{
PMinPMax = 4,10
MatchingIonTolerancePPM = 100.
}
let peptideSpectrumMatchingParams :Dto.PeptideSpectrumMatchingParams =
{
ChargeStateDeterminationParams = chargeDetermParams
LookUpPPM = 30.
nTerminalSeries = NTerminalSeries.B
cTerminalSeries = CTerminalSeries.Y
Andromeda = andromedaParams
}
let serialized =
peptideSpectrumMatchingParams
|> JsonConvert.SerializeObject
Executing the Tool
Disclaimer this tool needs a peptide database to query against, if you did not create one yet you can do so by using the PeptideDB tool.
To score all MS/MS of an MS run simply call:
proteomiqon-peptidespectrummatching -i "path/to/your/run.mzml" -d "path/to/your/database.sqlite" -o "path/to/your/outDirectory" -p "path/to/your/params.json"
It is also possible to call the tool on a list of MS files. If you have a mulitcore cpu it is possible to score multiple runs in parallel using the -c flag:
proteomiqon-peptidespectrummatching -i "path/to/your/run1.mzml" "path/to/your/run2.mzml" "path/to/your/run3.mzml" -d "path/to/your/database.sqlite" -o "path/to/your/outDirectory" -p "path/to/your/params.json" -c 3
A detailed description of the CLI arguments the tool expects can be obtained by calling the tool:
proteomiqon-peptidespectrummatching --help
from BioFSharp.Mz
from ProteomIQon
from BioFSharp.Mz
{ ExpectedMinimalCharge: int
ExpectedMaximumCharge: int
Width: float
MinIntensity: float
DeltaMinIntensity: float
NrOfRndSpectra: int }
{ PMinPMax: int * int
MatchingIonTolerancePPM: float }
from ProteomIQon
module PeptideSpectrumMatchingParams
from ProteomIQon.Dto
--------------------
type PeptideSpectrumMatchingParams =
{ ChargeStateDeterminationParams: ChargeDetermParams
LookUpPPM: float
nTerminalSeries: NTerminalSeries
cTerminalSeries: CTerminalSeries
Andromeda: AndromedaParams }
module NTerminalSeries
from ProteomIQon.Common
--------------------
type NTerminalSeries = (BioFSharp.IBioItem -> float) -> BioFSharp.AminoAcids.AminoAcid list -> PeakFamily<TaggedMass.TaggedMass> list
module CTerminalSeries
from ProteomIQon.Common
--------------------
type CTerminalSeries = (BioFSharp.IBioItem -> float) -> BioFSharp.AminoAcids.AminoAcid list -> PeakFamily<TaggedMass.TaggedMass> list
static member DeserializeAnonymousType<'T> : value: string * anonymousTypeObject: 'T -> 'T + 1 overload
static member DeserializeObject : value: string -> obj + 7 overloads
static member DeserializeXNode : value: string -> XDocument + 3 overloads
static member DeserializeXmlNode : value: string -> XmlDocument + 3 overloads
static member EnsureDecimalPlace : value: float * text: string -> string + 1 overload
static member EnsureFloatFormat : value: float * text: string * floatFormatHandling: FloatFormatHandling * quoteChar: char * nullable: bool -> string
static member PopulateObject : value: string * target: obj -> unit + 1 overload
static member SerializeObject : value: obj -> string + 7 overloads
static member SerializeObjectInternal : value: obj * type: Type * jsonSerializer: JsonSerializer -> string
static member SerializeXNode : node: XObject -> string + 2 overloads
...
JsonConvert.SerializeObject(value: obj, settings: JsonSerializerSettings) : string
JsonConvert.SerializeObject(value: obj, [<System.ParamArray>] converters: JsonConverter []) : string
JsonConvert.SerializeObject(value: obj, formatting: Formatting) : string
JsonConvert.SerializeObject(value: obj, formatting: Formatting, settings: JsonSerializerSettings) : string
JsonConvert.SerializeObject(value: obj, type: System.Type, settings: JsonSerializerSettings) : string
JsonConvert.SerializeObject(value: obj, formatting: Formatting, [<System.ParamArray>] converters: JsonConverter []) : string
JsonConvert.SerializeObject(value: obj, type: System.Type, formatting: Formatting, settings: JsonSerializerSettings) : string