![]()
![]()
![]()
Protein Inference
Disclaimer this tool needs a peptide database and peptide spectrum matches which passed fdr thresholds.
MS-based shotgun proteomics estimates protein abundances using a proxy: peptides. The process of 'Protein Inference' is concerned with the mapping of identified peptides to the proteins they putatively originated from. This process is not as straightforward as one might think at a first glance on the subject, since the peptide-to-protein mapping is not necessarily a one-to-one relationship but in many cases a one-to-many relationship. This is due to the fact that many proteins share peptides with an identical sequence, e.g. two proteins originating from two different splice variants of the same gene.
One way to cope with this problem is to introduce the concept of protein groups, which allow us to report the aggregation of all peptides which map to all isoforms of a gene independently from the peptides mapping uniquely to a single isoform. While this approach has its merits it leaves room for fine tuning when implemented. Lets say we have two proteins pA and pB which were both discovered by one peptide uniquely mapping to each of them and additionally by a third peptide, which maps to both of them: How do we report our findings? We could report both proteins seperately and as a protein group, we could only report the protein group, or we could report both proteins but not the protein group. A problem of comparable complexity occurs when we think about peptides when calculating the abundances for the proteingroup pA;pB. Do we use the peptides only once, or do we also use the peptides mapping uniquely to protein pA and pB? Fortunately, the tool ProteinInference gives you the possibility to choose any of the described scenarios by tuning the parameters described below. The following scheme gives an overview how parameter settings influence inferred protein groups:
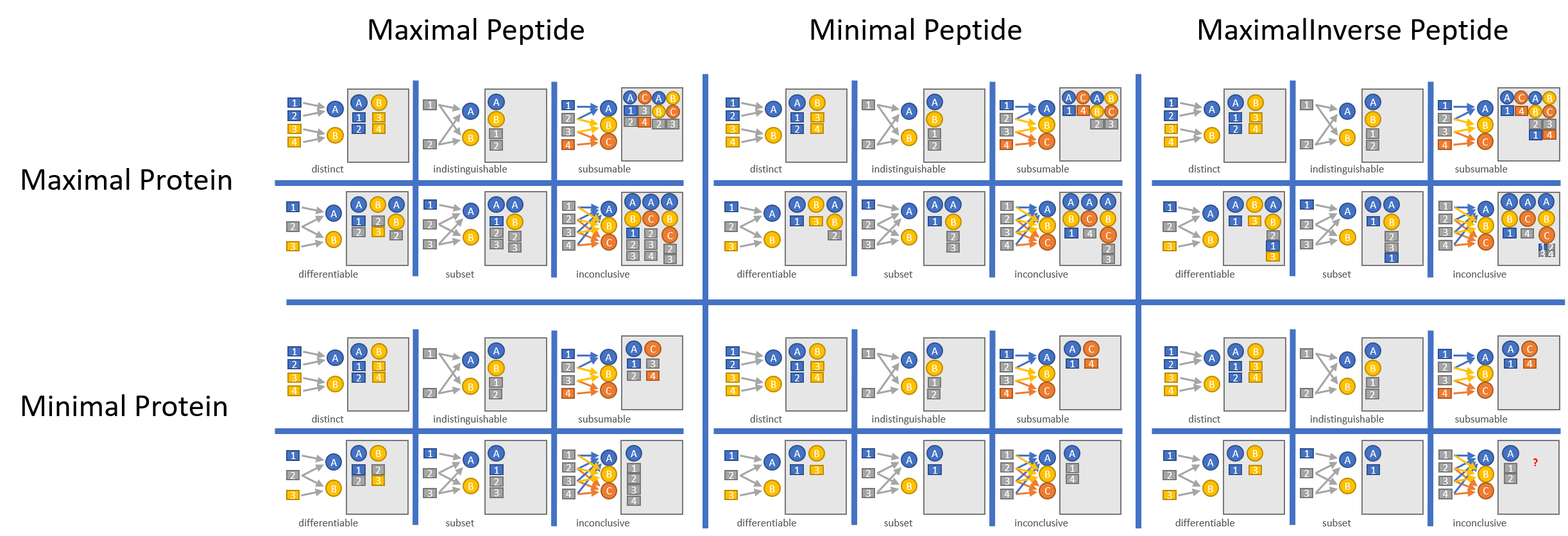
Moreover, we report each protein group with a so called 'Peptide evidence class'. This metric gives an indication how pure the peptide composition of a protein group is and lets us differentiate between protein groups that consist of isoforms of a splice variant or contain a rather arbitrary mix of proteins. In order to determine these inter-protein relationships the user can optionally supply a gff3 file.
Parameters
The following table gives an overview of the parameter set:
Parameter |
Default Value |
Description |
|---|---|---|
ProteinIdentifierRegex |
id |
Regex pattern for parsing of the protein IDs in the database |
Protein |
ProteinInference.IntegrationStrictness.Maximal |
Parameter to tune creation of protein groups (see scheme above) |
Peptide |
ProteinInference.PeptideUsageForQuantification.Minimal |
Parameters to tune the way peptides are aggregated (see scheme above) |
GroupFiles |
true |
Indicates if information is shared accross .qpsm files |
GetQValue |
QValueMethod.Storey |
Lets the user choose between published FDR calculation methods |
Parameter Generation
Parameters are handed to the cli tool as a .json file. you can download the default file here, or use an F# script, which can be downloaded or run in Binder at the top of the page, to write your own parameter file:
#r "nuget: ProteomIQon, 0.0.5"
#r "nuget: BioFSharp.Mz, 0.1.5-beta"
open ProteomIQon
open ProteomIQon.Domain
open BioFSharp.Mz
let defaultProteinInferenceParams: Dto.ProteinInferenceParams =
{
ProteinIdentifierRegex = "id"
Protein = ProteinInference.IntegrationStrictness.Maximal
Peptide = ProteinInference.PeptideUsageForQuantification.Minimal
GroupFiles = true
GetQValue = QValueMethod.Storey
}
let serialized =
defaultProteinInferenceParams
|> Json.serialize
Executing the Tool
Disclaimer this tool needs a peptide database and peptide spectrum matches which passed fdr thresholds.
To map all identified peptide sequences of an MS run to a protein group simply call:
proteomiqon-proteininference -i "path/to/your/run.qpsm" -d "path/to/your/database.sqlite" -g "path/to/your/proteom.gff3" -o "path/to/your/outDirectory" -p "path/to/your/params.json"
It is also possible to call the tool on a lists of scored psm files:
proteomiqon-proteininference -i "path/to/your/run1.qpsm" "path/to/your/run2.qpsm" "path/to/your/run3.qpsm" -d "path/to/your/database.sqlite" -g "path/to/your/proteom.gff3" -o "path/to/your/outDirectory" -p "path/to/your/params.json"
A detailed description of the CLI arguments the tool expects can be obtained by calling the tool:
proteomiqon-proteininference --help
from ProteomIQon
from ProteomIQon
module ProteinInferenceParams
from ProteomIQon.Dto
--------------------
type ProteinInferenceParams =
{ ProteinIdentifierRegex: string
Protein: IntegrationStrictness
Peptide: PeptideUsageForQuantification
GroupFiles: bool
GetQValue: QValueMethod }
from BioFSharp.Mz
| Minimal
| Maximal
| Minimal
| Maximal
| MaximalInverse
| Storey
| LogisticRegression of FDRMethod
| NoQValue
from ProteomIQon